Contributed by:
Shahnaz Khan, MPH
Vice President, Value & Access
RTI Health Solutions
Now is the time for drug manufacturers to do everything they can to get ready for a potential submission in support of their product for Medicare's Drug Price Negotiation (DPN) program. To fully prepare for a potential submission for the DPN, manufacturers can evaluate the chances that their product might be selected, determine what evidence Centers for Medicare and Medicaid Services (CMS) will most likely require, and learn how that evidence will be prioritized.
The goal of the US Inflation Reduction Act (IRA) program is to negotiate a maximum fair price (MFP) for certain high expenditures, single-source drugs, or biologic products that do not have generic or biosimilar competition. As the guidance is currently written, negotiations will last for 1 year and prices will become effective 2 years after that. The prices for the first round of 10 drugs chosen for negotiation in 2023 will become effective in 2026. The year the negotiated prices become effective is known as the Initial Price Applicability Year, or IPAY. CMS will select an additional 15 drugs in 2025 with an IPAY of 2027, followed by 15 additional Medicare Part D or Part B drugs for IPAY 2028. The plan is to choose 20 drugs for IPAY 2029 and each year into the future.
THE DRUG SELECTION PROCESS
The DPN program focuses on products that have been on the market for a long time and are the costliest from a Medicare perspective. In other words, the aim of DPN is to realize the greatest savings possible.
Initial Drug Selection Criteria for Initial Price Applicability Year 2027
- Drugs covered under Medicare Part D
- Drug products ≥ 7 years for small-molecule drugs or 11 years for biologics past their Food and Drug Administration (FDA) approval or licensure date; if a drug has multiple indications, the earliest approval date applies
- Delays in selection to the negotiation-eligible list are allowed for biological products where there is a “high likelihood” of biosimilar market entry within 2 years of the publication date of the selected drug list
- In the case of a fixed combination product with ≥ 2 active ingredients, the distinct combination of active ingredients will be considered as 1 active ingredient
Figure 1. Drug Selection Process for Initial Price Applicability Year 2027
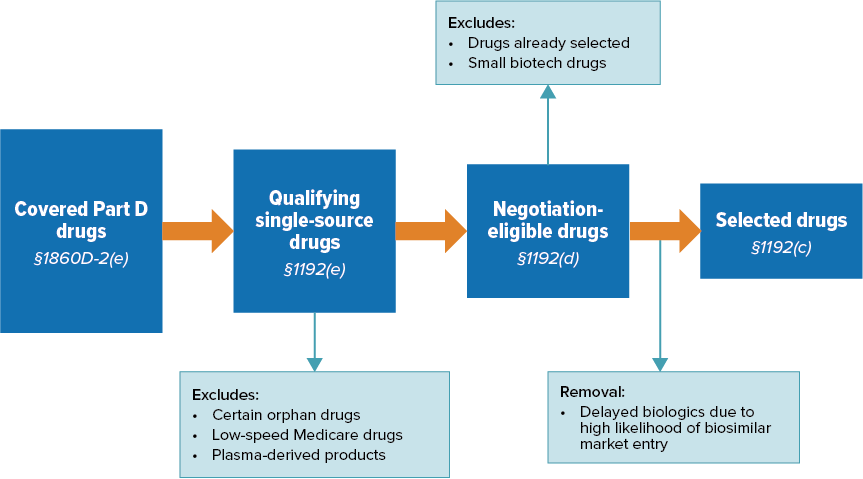
Source: CMS (2024).
Ranking
Once CMS identifies the 50 qualifying single-source drugs with the highest total expenditures under Part D:
- They will rank them by total expenditures under Medicare Part D, drugs with the highest total expenditures being listed first
- Next, CMS will remove any biologics that qualify for delayed selection from the list
- Finally, CMS will select the 15 highest ranked drugs and will publish this list by 1 February 2025
Excluded Drugs
- Orphan drugs designated and approved for only 1 rare disease or condition
- Drugs with total spending under Medicare Part D and Part B combined of less than $200 million
- Plasma-derived products
- Small biotech drugs (2026-2028)
Using the guidance on drug selection criteria, manufacturers can identify if their product(s) are likely to be selected as one of the products for IPAY 2027. This will allow for the preparation of data submissions to CMS well in advance of the selection announcement in February 2025.
WHAT MATERIALS WILL THE CENTERS FOR MEDICARE AND MEDICAID SERVICES REQUEST?
Information from Manufacturers
- Manufacturer research and development costs and the extent to which the manufacturer has recouped these costs
- Current unit costs of production and distribution
- Federal financial support for novel therapeutic discovery and development
- Data on pending and approved patent applications, exclusivities, and certain other applications and approvals
- Market data and revenue and sales volume data in the US
There is, however, no specific information on how CMS will weigh these different elements when calculating an MFP, which leaves some wondering how useful submitted information will be. Manufacturers must provide the above data by 1 March 2025. The takeaway from this is that research and development costs, along with sales and revenue data, should be readily available ahead of time so manufacturers can compile them for a timely submission in the case that the drug is selected.
Information About Therapeutic Alternatives
CMS will be looking for:
- How advanced the selected drug is when compared with existing alternatives and their costs
- Prescribing information for the selected drug and its therapeutic alternatives (which may include generics or biosimilars)
- Comparative effectiveness of the selected drug and its therapeutic alternatives, including their effects on specific populations (e.g., individuals with disabilities, older adults, terminally ill individuals, children)
- Note here that the guidance states, “In considering impact on specific populations and patients with unmet medical needs, CMS will prioritize research specifically designed to focus on those populations”
- How well the selected drug and its alternatives address unmet needs for a condition that is not adequately addressed by available therapy
Sources for Identifying Therapeutic Alternatives
CMS will determine therapeutic alternatives for the indications of a selected drug using data submitted by the manufacturer and the public (this can include other drug manufacturers, people with Medicare, clinicians, and others). The CMS will also consider information from these sources:
- FDA-approved indications
- Drug classification systems commonly used for formulary development
- CMS-recognized Part D compendia
- Widely accepted clinical guidelines
- A CMS-led literature review
- Drug or drug class reviews
- Peer-reviewed studies
When there are many potential therapeutic alternatives for a given indication of the selected drug, CMS may focus on a subset of therapeutic alternatives that are clinically comparable with the selected drug for the purpose of developing the initial offer.
Key Takeaways
In addition, CMS states that it will review real-world evidence (RWE), conduct an internal analysis, and consult with experts regarding evidence of the clinical benefits of the selected drugs and their therapeutic alternatives. Note that CMS will only consider therapeutic alternatives that are drugs or biologic products (including generics and biosimilars) and not nonpharmaceutical alternatives.
Manufacturers should carefully consider therapeutic alternatives to their product(s) and understand how to narrow the list if there are many alternatives. Once the list of therapeutic alternatives is developed, a literature review and qualitative synthesis will be needed to gather information on the therapeutic alternatives and relevant outcomes. The guidance is not clear regarding the need for quantitative analyses such as an indirect treatment comparison. Companies can work with their research partners to carefully consider whether the evidence may lend itself to potential quantitative analyses. As a first step, companies can perform a feasibility analysis.
COMPARING RELEVANT OUTCOMES
When comparing the selected drug to its therapeutic alternatives, CMS will define relevant outcomes based on a CMS-led literature review as well as the information submitted by manufacturers and other sources (e.g., patients and caregivers). The following outcomes may be considered relevant in determining clinical benefit:
- Cure, survival, progression-free survival, or improved morbidity
- Changes in symptoms or other factors that are of importance to patients and patient-reported outcomes
- Changes to productivity, independence, and quality of life to the extent that these outcomes correspond with a direct impact on individuals taking the drug, including patient-centered outcomes when available
- Caregiver perspective to the extent that it reflects directly upon the experience or relevant outcomes of the patient taking the selected drug
Throughout the life cycle of the product, the RWE strategy becomes more crucial for products expected to be subject to the IRA because RWE can fill in gaps in evidence and strengthen the overall body of evidence regarding safety, effectiveness, and overall value of a product.
Per the IRA, CMS “shall not use evidence from comparative clinical-effectiveness research in a manner that treats extending the life of an elderly, disabled, or terminally ill individual as of lower value than extending the life of an individual who is younger, non-disabled, or not terminally ill”—i.e., the use of quality-adjusted life-years (QALYs) in not permitted. From the perspective of CMS, the use of QALYs essentially devalues the life of specific members when determining cost-effectiveness of a treatment and is therefore inherently discriminatory toward these populations.
Potential alternatives to QALYs include equal value of life-years gained (evLYG), Healthy Years in Total (HYT), and the Generalized Risk-Adjusted Cost-Effectiveness (GRACE) approach. However, HR 485, which proposed banning “other similar metrics” in federal healthcare program determinations, passed in the US House in February 2024. New laws around the use of QALY alternatives may potentially restrict cost-effectiveness analyses in IRA negotiations.
DATA COLLECTION AND SUBMISSION
During the summer of 2024, CMS intends to publish a revised Information Collection Request (ICR) for IPAY 2027, with a 60-day public comment period. The ICR will describe how CMS will collect the relevant data and provide instructions on how manufacturers and the public may submit such data. CMS will likely group questions into the following categories:
- Manufacturer input, including information on FDA-approved indications and off-label uses of the product listed in CMS-recognized Part D compendia
- Patient or caregiver experience, including factors a patient considers most important when assessing the drug’s value
- Clinical experience
- Health research (e.g., economic and health equity data)
Types of Evidence Center for Medicare and Medicaid Services Will Consider
- Literature reviews and RWE, including manufacturer-conducted literature reviews
- Analytics conducted by CMS (e.g., CMS can use their own claims data to determine indications of focus)
- Subject matter and clinical expert consultations
- Evidence related specifically to Medicare populations, including on individuals with disabilities, patients with end-stage renal disease, and Medicare-aged populations
How Will Evidence Be Prioritized?
In reviewing the evidence, CMS will consider:
- Source
- Rigor of the study methodology
- Current relevance to the selected drug and its therapeutic alternative(s)
- Whether the study has been through peer review
- Study limitations
- Degree of certainty regarding conclusions
- Risk of bias
- Study time horizons
- Generalizability
- Study population
- Relevance to negotiation factors
The key is that the CMS will prioritize research evidence from rigorously conducted studies. When conducting literature reviews to gather evidence, manufacturers should focus on evidence from rigorous observational studies and randomized controlled trials over smaller, nonrandomized trials when available. This is true particularly for studies conducted in special populations such as Medicare populations, or that define unmet needs.
What is the bottom line for manufacturers? Be part of the process, be ready for what is coming, and understand whether you have the capacity to identify and synthesize key evidence yourself or if you might need to find a partner who can help you prepare and walk you through the process.
TIMELINE FOR INITIAL PRICE APPLICABILITY YEAR 2027
Once a drug product is selected, the manufacturer will have approximately 1 month to submit specific economic and market data for the selected product(s) as well as information related to therapeutic alternatives, unmet needs, and impacts on specific populations. The negotiations will take place from June-November 2025, with a deadline of 30 November 2025 for CMS to publish the negotiated MFPs. The negotiated MFPs will take effect 1 January 2027. The timeline below details the next steps for the drugs that have been chosen.
1 March 2025: Deadline for Manufacturer and Public Data Submissions
Manufacturers of drugs selected in Round 2 must submit specific economic and market data to CMS (e.g., research and development costs, costs of production, and sales and revenue data). The public may submit data on therapeutic alternatives to the selected drugs, data related to unmet medical needs, and data on impacts to specific populations among other considerations
This deadline is the day after CMS publishes an explanation of Round 1 negotiations. Some have suggested that CMS delay releasing the list for Round 2 to allow manufacturers to review the explanations for Round 1 and learn from them
Spring 2025: Public and Manufacturer Engagement
CMS will provide public engagement opportunities for patients, providers, advocacy groups, and other interested parties. CMS will also provide an engagement opportunity for the manufacturers of selected drugs to meet with CMS to discuss data submission
1 June 2025: Initial Offer From CMS
Deadline for CMS to submit initial offer for MFP for a selected drug, with a concise justification to each drug’s manufacturer
1 July 2025: Manufacturer Response Deadline
Deadline for manufacturers of selected drugs to respond to the initial MFP offer
Summer 2025: Negotiation Period
31 October 2025: Manufacturer Acceptance Deadline
Deadline for manufacturers of selected drugs to accept or reject final offer for MFP
1 November 2025: End of Negotiation Period
30 November 2025: Deadline for CMS to publish negotiated MFPs from Round 2
1 March 2026: CMS Publication of MFPs
Deadline for CMS to publish an explanation of MFPs for Round 2, including a narrative explanation of the negotiation process and certain additional information
1 January 2027: Negotiated MFPs for Round 2 Drugs Go Into Effect