Article contributed by: Shahnaz Khan, MPH, Executive Director, Market Access and Outcomes Strategy at RTI Health Solutions
FDA Guidance
In June 2018, the United States Food and Drug Administration (US FDA) released an important guidance document – “Drug and Device Manufacturer Communications with Payors, Formulary Committees, and Similar Entities–Questions and Answers.” The document addresses frequently asked questions about health care economic information for approved products. It also addresses key questions about the communication of information for unapproved products as well as unapproved uses of approved products.
Payors prefer to receive information from manufacturers well in advance of a product’s approval by the FDA so that they can make timely coverage and reimbursement decisions. The guidance provides recommendations for those communications.
In summary, the guidance recommends providing the following unbiased, factual, accurate, and non-misleading information.
- Information about the indications sought
- The anticipated timeline for possible FDA approval, clearance, or licensure of the product or new use
- Product pricing information
- Patient utilization projections (e.g., epidemiological data projection on incidence and prevalence)
- Product-related programs or services
- Factual presentations of results from studies, including clinical studies (i.e., no characterizations or conclusions regarding the safety or effectiveness of the unapproved product or the unapproved use)
In addition to the above information, the guidance recommends including the following in the pre-approval document.
- A clear statement that the product or use is not approved, cleared, or licensed, and that the safety or effectiveness of the product or use has not been established
- Information related to the stage of product development (e.g., the status of any study in which a product or new use is being investigated, how it relates to the overall product development plan, whether a marketing application for the product or new use has been submitted to FDA, or when submission is planned)
- For communications that include factual presentations of results from studies, a description of material aspects of study design and methodology, and disclosure of material limitations related to design, methodology, and results
- Inclusively presented results (i.e., both positive and negative or null findings should be presented)
- A prominent statement disclosing FDA approved, cleared, or licensed indications and a copy of the most current FDA-required labeling
- As new information or updated versions of previously communicated information regarding a product become available, the FDA guidance recommends providing this information to the payor
AMCP Format for Formulary Submissions Guidance
Version 4.0 (April 2016)
The AMCP Format v4.0 states that manufacturers may use the pre-approval submissions guidance as a template for information in response to a payor’s request for a pre-approval “dossier.” Include these elements:
- Clinical trial information from Phase 1, Phase 2, and Phase 3 studies (peer-reviewed publications; congress abstracts, posters, and presentations; the company’s internal standard response letters; or clinicaltrials.gov information)
- Pre-clinical studies
- Unpublished data on file – at the discretion of the manufacturer
- Disease information
- Product information
Version 4.1 (2020)
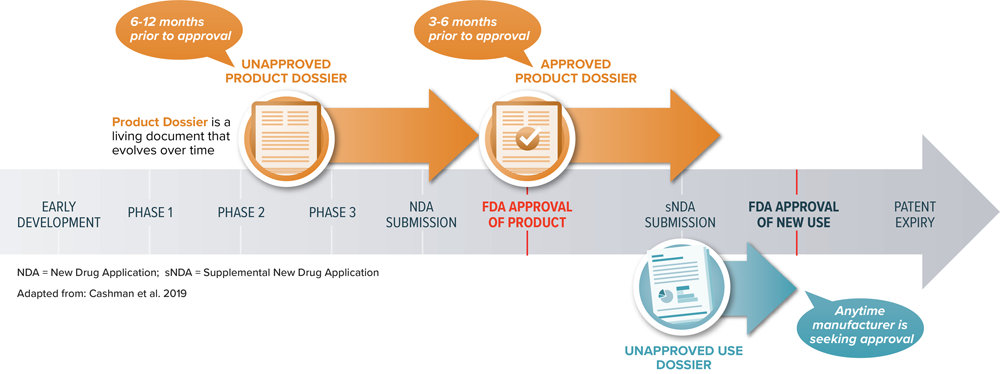
At the October 2019 AMCP Nexus meeting, the AMCP Format Executive Committee (FEC) provided an overview of upcoming changes to the AMCP guidelines. It is expected that these changes will align AMCP guidance more closely with FDA guidance. This will be the sixth update to the Format. The FEC outlined three distinct dossier types that will be noted:
- Unapproved Product Dossier: to communicate information about an unapproved product for which initial FDA approval is being sought
- Approved Product Dossier: to communicate information (clinical and economic evidence) about an approved product; provided based on an unsolicited request
- Unapproved Use Dossier: to communicate information about an unapproved use (of an approved product) for which FDA approval is being sought
Important Considerations
- An unapproved-product dossier would eventually be converted into the approved product dossier as the product nears approval. These documents should not co-exist.
- The unapproved-use dossier is a separated document that may co-exist with a product dossier.
- Regardless of dossier type, the dossier should be presented to payors by staff with appropriate medical and scientific credentials and applicable responsibilities.
AMCP FEC Recommendations for Content of Each Dossier (Key Sections)
| Unapproved Product Dossier | Approved Product Dossier | Unapproved Use Dossier | |
|---|---|---|---|
| Section 1 | Highlights and Overview in a single table (no executive summary/value proposition) | Executive Summary highlighting clinical and economic value | Highlights and Overview in a single table (no executive summary/value proposition) |
| Section 2 | Product Information and Disease Description | ||
| Section 3 | Clinical Evidence - studies supporting unapproved product; may be text and evidence tables | Clinical Evidence - approved indications as well as off-label uses; text and evidence tables | Clinical Evidence - studies supporting unapproved use; may be text and evidence tables |
| Section 4 | Economic Information - provide as much pricing information as possible; CEA and BIM likely not possible | Economic Value and Modeling Report - cost-effectiveness model and BIM | Economic Information - pricing should be available since product approved; economic models likely not possible |